
Carbanion - Carbanion
Un carbanion est un anion dans lequel le carbone est trivalent (formant trois liaisons) et porte une charge négative formelle (sous au moins une forme de résonance significative).
Formellement, un carbanion est la base conjuguée d'un acide carboné :
- R 3 CH + :B − → R 3 C : − + HB
où B représente la base. Les carbanions formés à partir de la déprotonation d'alcanes (à un carbone sp 3 ), d'alcènes (à un carbone sp 2 ), d'arènes (à un carbone sp 2 ) et d'alcynes (à un carbone sp) sont appelés alkyle , alcényle (vinyle) , aryle et alcynyle (acétylure) anions , respectivement.
Les carbanions ont une concentration de densité électronique au niveau du carbone chargé négativement, qui, dans la plupart des cas, réagit efficacement avec une variété d' électrophiles de forces variables, y compris les groupes carbonyle , les imines / sels d'iminium , les réactifs d'halogénation (par exemple, N- bromosuccinimide et diiode ) , et les donneurs de protons . Un carbanion est l'un des nombreux intermédiaires réactifs de la chimie organique . En synthèse organique, les réactifs organolithiens et les réactifs de Grignard sont couramment traités et appelés « carbanions ». Il s'agit d'une approximation pratique, bien que ces espèces soient généralement des amas ou des complexes contenant des liaisons très polaires, mais toujours covalentes, des liaisons métal-carbone (M + –C δ− ) plutôt que de véritables carbanions.
Géométrie
En l'absence de délocalisation , la charge négative d'un carbanion est localisée dans une orbitale sp x hybridée sur carbone comme une paire isolée . En conséquence, les carbanions alkyle, alcényle/aryle et alcynyle localisés prennent respectivement des géométries trigonales pyramidales, courbées et linéaires. Selon la règle de Bent , le placement des électrons de la paire solitaire carbanionique dans une orbitale avec un caractère s significatif est favorable, ce qui explique les géométries pyramidalisées et courbées des carbanions alkyle et alcényle, respectivement. La théorie de la répulsion des paires d'électrons en coquille de Valence (VSEPR) fait des prédictions similaires. Cela contraste avec les carbocations, qui ont une préférence pour les orbitales non-liantes inoccupées de caractère p atomique pur, conduisant à des géométries planes et linéaires, respectivement, pour les carbocations alkyle et alcényle.


Cependant, les carbanions délocalisés peuvent s'écarter de ces géométries. Au lieu de résider dans une orbitale hybride, la paire solitaire carbanionique peut à la place occuper une orbitale p (ou une orbitale de caractère p élevé). Une orbitale p a une forme et une orientation plus appropriées pour se chevaucher avec le système π voisin, ce qui entraîne une délocalisation de charge plus efficace. En conséquence, les carbanions d'alkyle avec des groupes de conjugaison voisins (par exemple, les anions allyliques, les énolates, les nitronates, etc.) sont généralement plans plutôt que pyramidaux. De même, les carbanions alcényles délocalisés favorisent parfois une géométrie linéaire plutôt que courbée. Le plus souvent, une géométrie courbée est toujours préférée pour les anions alcényles substitués, bien que la géométrie linéaire ne soit que légèrement moins stable, ce qui permet un équilibrage facile entre les isomères ( E ) et ( Z ) de l'anion (courbé) à travers un état de transition linéaire. Par exemple, les calculs indiquent que l'anion vinyle parent, H 2 C=CH − , a une barrière d'inversion de 27 kcal/mol (110 kJ/mol), tandis que l'anion allényle, H 2 C=C=H (↔ H 2–C≡CH), dont la charge négative est stabilisée par délocalisation, a une barrière d'inversion de seulement 4 kcal/mol (17 kJ/mol), traduisant une stabilisation de l'état de transition linéaire par une meilleure délocalisation π.
Tendances et occurrence
Les carbanions sont typiquement nucléophiles et basiques. La basicité et la nucléophilie des carbanions sont déterminées par les substituants sur le carbone. Ceux-ci inclus
- l' effet inductif . Les atomes électronégatifs adjacents à la charge stabiliseront la charge;
- le degré de conjugaison de l'anion. Les effets de résonance peuvent stabiliser l'anion. Cela est particulièrement vrai lorsque l'anion est stabilisé en raison de l' aromaticité .
La géométrie affecte également l' hybridation orbitale du carbanion porteur. Plus le caractère s de l'atome porteur de charge est grand, plus l'anion est stable.
Les réactifs organométalliques comme le butyllithium (groupe hexamérique, [BuLi] 6 ) ou le bromure de méthylmagnésium (complexe d'éther, MeMgBr(OEt) 2 ) sont souvent appelés « carbanions », au moins dans un sens rétrosynthétique . Cependant, ce sont vraiment des amas ou des complexes contenant une liaison covalente polaire, mais avec une densité électronique fortement polarisée vers l'atome de carbone. En effet, les vrais carbanions sans substituants stabilisants ne sont pas disponibles en phase condensée, et ces espèces doivent être étudiées en phase gazeuse.
Pendant un certain temps, on ne savait pas si de simples anions alkyles pouvaient exister en tant qu'espèces libres ; de nombreuses études théoriques ont prédit que même l' anion méthanide CH–
3devrait être une espèce non liée (c'est-à-dire l' affinité électronique de CH•
3était prédit négatif). Une telle espèce se décomposerait immédiatement par éjection spontanée d'un électron et serait donc trop fugace pour être observée directement par spectrométrie de masse. Cependant, en 1978, l'anion méthanide a été synthétisé sans ambiguïté en soumettant le cétène à une décharge électrique, et l'affinité électronique (EA) de CH•
3a été déterminé par spectroscopie photoélectronique à +1,8 kcal/mol, ce qui en fait une espèce liée, mais à peine. La structure de CH–
3s'est avéré pyramidal (C 3v ) avec un angle H−C−H de 108° et une barrière d' inversion de 1,3 kcal/mol, tandis que CH•
3a été déterminé comme étant plan ( groupe de points D 3h ).
Carbanions simples primaires, secondaires et tertiaires sp 3 (par exemple, éthanide CH
3CH–
2, isopropanide (CH
3)
2CH−
, et le t -butanide (CH 3 ) 3 C − ) ont ensuite été déterminés comme étant des espèces non liées (les EA de CH 3 CH•
2, (CH 3 ) 2 CH • , (CH 3 ) 3 C • sont -6, -7,4, -3,6 kcal/mol, respectivement) indiquant que la substitution est déstabilisante. Cependant, des effets stabilisateurs relativement modestes peuvent les rendre liés. Par exemple, les anions cyclopropyle et cubyle sont liés en raison de l'augmentation du caractère s de l'orbitale à paire isolée, tandis que les anions néopentyle et phénéthyle sont également liés, en raison de l'hyperconjugaison négative de la paire isolée avec le substituant (n C → σ * C–C ). Il en va de même pour les anions à stabilisation benzylique et allylique . Les carbanions en phase gazeuse qui sont hybrides sp 2 et sp sont beaucoup plus fortement stabilisés et sont souvent préparés directement par déprotonation en phase gazeuse.
Dans la phase condensée, seuls les carbanions suffisamment stabilisés par délocalisation ont été isolés en tant qu'espèces véritablement ioniques. En 1984, Olmstead et Power ont présenté le sel d' éther couronne de lithium du carbanion de triphénylméthanide issu du triphénylméthane , du n- butyllithium et du 12-couronne-4 (qui forme un complexe stable avec les cations lithium) à basse température :

L'ajout de n- butyllithium au triphénylméthane (p K a dans le DMSO de CHPh 3 = 30,6) dans le THF à basse température suivi de 12-couronne-4 donne une solution rouge et le complexe salin [Li(12-couronne-4)] + [CPh 3 ] − précipite à −20 °C. Les longueurs de liaison C-C centrales sont de 145 pm avec le cycle phényle propulsé à un angle moyen de 31,2°. Cette forme d'hélice est moins prononcée avec un contre-ion tétraméthylammonium. Une structure cristalline pour l'anion diphénylméthanide analogue ([Li(12-crown-4)] + [CHPh 2 ] − ), préparé à partir de diphénylméthane (p K a dans le DMSO de CH 2 Ph 2 = 32,3), a également été obtenue. Cependant, la tentative d'isolement d'un complexe de l'anion benzylique [CH 2 Ph] − du toluène (p K a dans le DMSO de CH 3 Ph ≈ 43) a échoué, en raison de la réaction rapide de l'anion formé avec le solvant THF. L'anion benzyle libre a également été généré dans la phase de solution par radiolyse pulsée du dibenzylmercure.
Au début de 1904 et 1917, Schlenk a préparé deux sels de couleur rouge, formulés respectivement comme [NMe 4 ] + [CPh 3 ] − et [NMe 4 ] + [CH 2 Ph] − , par métathèse du réactif organosodium correspondant avec du tétraméthylammonium chlorure. Étant donné que les cations tétraméthylammonium ne peuvent pas former de liaison chimique avec le centre carbanionique, on pense que ces espèces contiennent des carbanions libres. Alors que la structure de la première a été vérifiée par cristallographie aux rayons X près d'un siècle plus tard, l'instabilité de la seconde a jusqu'à présent exclu la vérification structurelle. Il a été rapporté que la réaction du putatif "[NMe 4 ] + [CH 2 Ph] − " avec de l'eau libérait du toluène et de l'hydroxyde de tétraméthylammonium et fournit une preuve indirecte de la formulation revendiquée.
Un outil pour la détection des carbanions en solution est la RMN du proton . Un spectre de cyclopentadiène dans le DMSO montre quatre protons vinyliques à 6,5 ppm et deux protons de pont méthylène à 3 ppm alors que l' anion cyclopentadiényle a une seule résonance à 5,50 ppm. L'utilisation de RMN 6 Li et 7 Li a fourni des données structurelles et de réactivité pour une variété d' espèces d' organolithium .
Acides carbonés
Tout composé contenant de l'hydrogène peut, en principe, subir une déprotonation pour former sa base conjuguée. Un composé est un acide carboné si la déprotonation entraîne la perte d'un proton d'un atome de carbone. Par rapport aux composés généralement considérés comme des acides (par exemple, les acides minéraux comme l'acide nitrique ou les acides carboxyliques comme l'acide acétique ), les acides carbonés sont généralement plus faibles de plusieurs ordres de grandeur, bien qu'il existe des exceptions (voir ci-dessous). Par exemple, le benzène n'est pas un acide au sens classique d' Arrhenius , puisque ses solutions aqueuses sont neutres. Néanmoins, il est très faiblement acide de Bronsted avec une estimation de p K a de 49 qui peut subir une déprotonation en présence d'une superbase comme la base de Lochmann-Schlosser ( n - butyl - lithium et de potassium t - butoxyde ). En tant que paires acide-base conjuguées, les facteurs qui déterminent la stabilité relative des carbanions déterminent également l'ordre des valeurs p K a des acides carbonés correspondants. De plus, les valeurs de p K a permettent de prédire si un processus de transfert de protons sera thermodynamiquement favorable : Pour que la déprotonation d'une espèce acide HA avec la base B − soit thermodynamiquement favorable ( K > 1), la relation p K a ( BH) > p K a (AH) doit être vérifié.
Ces valeurs ci-dessous sont des valeurs p K a déterminées dans le diméthylsulfoxyde (DMSO), qui a une plage utile plus large (~0 à ~35) que les valeurs déterminées dans l'eau (~0 à ~14) et reflètent mieux la basicité des carbanions dans des solvants organiques. Les valeurs inférieures à 0 ou supérieures à 35 sont estimées indirectement ; par conséquent, la précision numérique de ces valeurs est limitée. Aqueuse p K a des valeurs sont également un phénomène courant dans la littérature, en particulier dans le contexte de la biochimie et de l' enzymologie. De plus, les valeurs aqueuses sont souvent données dans les manuels d'introduction à la chimie organique pour des raisons pédagogiques, bien que la question de la dépendance aux solvants soit souvent passée sous silence. En général, les valeurs de p K a dans l'eau et le solvant organique divergent de manière significative lorsque l'anion est capable de liaison hydrogène. Par exemple, dans le cas de l'eau, les valeurs diffèrent considérablement : le p K a dans l'eau de l'eau est de 14,0, tandis que le p K a dans le DMSO de l'eau est de 31,4, ce qui reflète la capacité différente de l'eau et du DMSO à stabiliser l' anion hydroxyde. . En revanche, pour le cyclopentadiène , les valeurs numériques sont comparables : le p K a dans l'eau est de 15, tandis que le p K a dans le DMSO est de 18.
Acidités carbonées par p K a dans le DMSO .
Ces valeurs peuvent différer de manière significative de p aqueuse K a les valeurs.Nom Formule Formule structurelle p K a dans le DMSO Cyclohexane C 6 H 12 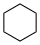
~60 Méthane CH 4 
~56 Benzène C 6 H 6 
~49 Propène C 3 H 6 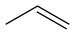
~44 Toluène C 6 H 5 CH 3 
~43 Ammoniac (N–H) NH 3 
~41 Dithiane C 4 H 8 S 2 
~39 Diméthylsulfoxyde (CH 3 ) 2 SO 
35,1 Diphénylméthane C 13 H 12 
32.3 Acétonitrile CH 3 CN 
31,3 Aniline (N–H) C 6 H 5 NH 2 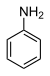
30.6 Triphénylméthane C 19 H 16 
30.6 Fluoroforme CHF 3 
30,5 Xanthène C 13 H 10 O 
30,0 Éthanol (O–H) C 2 H 5 OH 
29,8 Phénylacétylène C 8 H 6 
28,8 Thioxanthène C 13 H 10 S 
28,6 Acétone C 3 H 6 O 
26,5 Chloroforme CHCl 3 
24,4 Benzoxazole C 7 H 5 NON 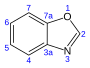
24,4 Fluorène C 13 H 10 
22,6 Indene C 9 H 8 
20.1 Cyclopentadiène C 5 H 6 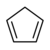
18,0 Nitrométhane CH 3 NON 2 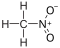
17.2 Malonate de diéthyle C 7 H 12 O 4 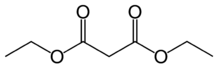
16.4 Acétylacétone (H 3 CCO) 2 CH 2 
13.3 Cyanure d'hydrogène HCN 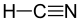
12.9 Acide acétique (O–H) CH 3 COOH 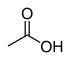
12.6 Malononitrile C 3 H 2 N 2 
11.1 Dimédone C 8 H 12 O 2 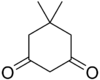
10.3 Acide de Meldrum C 6 H 8 O 4 
7.3 Hexafluoroacétylacétone (F 3 CCO) 2 CH 2 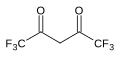
2.3 Chlorure d'hydrogène (Cl–H) HCl HCl (g) -2,0 Acide triflidique HC(SO 2 CF 3 ) 3 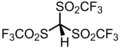
~ −16





















- Notez que l'acide acétique, l'ammoniac, l'aniline, l'éthanol et le chlorure d'hydrogène ne sont pas des acides carbonés, mais sont des acides communs présentés à titre de comparaison.
Comme l'indiquent les exemples ci-dessus, l'acidité augmente (p K a diminue) lorsque la charge négative est délocalisée. Cet effet se produit lorsque les substituants sur le carbanion sont insaturés et/ou électronégatifs. Bien que les acides carbonés soient généralement considérés comme des acides beaucoup plus faibles que les acides de Brønsted "classiques" comme l'acide acétique ou le phénol, l'effet cumulatif (additif) de plusieurs substituants accepteurs d'électrons peut conduire à des acides aussi forts ou plus forts que le minéral inorganique. acides. Par exemple, le trinitrométhane HC(NO 2 ) 3 , le tricyanométhane HC(CN) 3 , le pentacyanocyclopentadiène C 5 (CN) 5 H et l' acide fulminique HCNO sont tous des acides forts avec des valeurs aqueuses de p K a qui indiquent un transfert de protons complet ou presque complet arroser. Acide Triflidic , avec trois fortement attracteur d' électrons triflyle groupes, p a une estimation K un bien en dessous de -10 ° C. À l'autre extrémité de l'échelle, on pense que les hydrocarbures ne portant que des groupes alkyle ont des valeurs de p K a comprises entre 55 et 65. La gamme des constantes de dissociation des acides pour les acides carbonés s'étend ainsi sur 70 ordres de grandeur.
L'acidité de l'hydrogène dans les composés carbonylés permet à ces composés de participer à des réactions synthétiques importantes de formation de liaisons C-C, y compris la réaction d'aldolisation et l' addition de Michael .
Carbanions chiraux
Avec la géométrie moléculaire d'un carbanion décrite comme une pyramide trigonale, la question est de savoir si les carbanions peuvent ou non afficher une chiralité , car si la barrière d'activation pour l'inversion de cette géométrie est trop faible, toute tentative d'introduction de la chiralité se terminera par une racémisation , similaire à l' azote. renversement . Cependant, il existe des preuves solides que les carbanions peuvent en effet être chiraux par exemple dans les recherches menées avec certains composés organolithiens .
La première preuve de l'existence de composés organolithiens chiraux a été obtenue en 1950. La réaction du 2-iodooctane chiral avec le s- butyllithium dans l'éther de pétrole à -70 °C suivie d'une réaction avec de la neige carbonique a donné principalement de l'acide 2-méthylbutyrique racémique mais aussi un quantité d' acide 2-méthyloctanoïque optiquement actif , qui n'a pu se former qu'à partir du 2-méthylheptyllithium également optiquement actif avec l'atome de carbone lié au lithium le carbanion :

En chauffant la réaction à 0 °C, l'activité optique est perdue. D'autres preuves ont suivi dans les années 1960. Une réaction de l' isomère cis du bromure de 2-méthylcyclopropyle avec du s- butyllithium à nouveau suivie d'une carboxylation avec de la neige carbonique a donné l' acide cis -2-méthylcyclopropylcarboxylique. La formation de l' isomère trans aurait indiqué que le carbanion intermédiaire était instable.

De la même manière, la réaction du (+)-( S )- l -bromo- l -méthyl-2,2-diphénylcyclopropane avec le n- butyllithium suivie d'une trempe avec du méthanol a donné un produit avec rétention de configuration :

Les composés chiraux de méthyllithium sont de date récente :
![Oxy[2H1]méthyllithiums chiraux. Bu signifie butyle, i-Pr signifie isopropyle.](https://wikipedia.org/wiki/File:PhosphatePhosphonateRearrangement.png)
Le phosphate 1 contient un groupe chiral avec un hydrogène et un substituant deutérium . Le groupe stannyle est remplacé par le lithium à l'intermédiaire 2 qui subit un réarrangement phosphate-phosphorane en phosphorane 3 qui, par réaction avec l'acide acétique, donne l' alcool 4 . Encore une fois dans la plage de -78 °C à 0 °C, la chiralité est préservée dans cette séquence de réaction. (L' énantiosélectivité a été déterminée par spectroscopie RMN après dérivatisation avec l'acide de Mosher .)
Histoire
Une structure carbanionique est apparue pour la première fois dans le mécanisme réactionnel de condensation du benjoin, comme proposé à juste titre par Clarke et Arthur Lapworth en 1907. En 1904, Wilhelm Schlenk a préparé Ph 3 C − NMe+
4dans une recherche d'azote pentavalent ( à partir de chlorure de tétraméthylammonium et
Ph 3 CNa ) et en 1914 , il a montré comment radicaux triarylméthyle pourraient être réduits à carbanions par les métaux alcalins L'expression carbanion a été introduit par Wallis et Adams en 1933 en tant que contrepartie chargée négativement de la carbonium ion