
Adénome hypophysaire - Pituitary adenoma
| Adénome hypophysaire | |
|---|---|
 | |
| Perte du champ visuel dans l' hémianopsie bitemporale : perte de vision périphérique affectant les deux yeux, résultant d'une tumeur-typiquement un adénome hypophysaire-exerçant une pression sur le chiasma optique . | |
| Spécialité | Oncologie , endocrinologie |
Les adénomes hypophysaires sont des tumeurs qui surviennent dans l' hypophyse . Les adénomes hypophysaires sont généralement divisés en trois catégories en fonction de leur fonctionnement biologique : les adénomes bénins, les adénomes invasifs et les carcinomes . La plupart des adénomes sont bénins, environ 35 % sont invasifs et seulement 0,1 % à 0,2 % sont des carcinomes. Les adénomes hypophysaires représentent de 10 à 25 % de toutes les tumeurs intracrâniennes et le taux de prévalence estimé dans la population générale est d'environ 17 %.
Les adénomes hypophysaires non invasifs et non sécrétants sont considérés comme bénins au sens littéral comme au sens clinique ; cependant, une méta-analyse récente (Fernández-Balsells, et al. 2011) des recherches disponibles a montré qu'il existe à ce jour peu d'études - de mauvaise qualité - pour soutenir ou réfuter cette hypothèse.
Les adénomes dépassant 10 mm (0,39 in) de taille sont définis comme des macroadénomes , ceux de moins de 10 mm (0,39 in) étant appelés microadénomes . La plupart des adénomes hypophysaires sont des microadénomes et ont une prévalence estimée à 16,7 % (14,4 % dans les études d' autopsie et 22,5 % dans les études radiologiques ). La majorité des microadénomes hypophysaires restent souvent non diagnostiqués, et ceux qui sont diagnostiqués sont souvent trouvés comme une découverte fortuite et sont appelés incidentalomes .
Les macroadénomes hypophysaires sont la cause la plus fréquente d' hypopituitarisme .
Alors que les adénomes hypophysaires sont fréquents, affectant environ un sur 6 de la population générale, les adénomes hypophysaires cliniquement actifs qui nécessitent un traitement chirurgical sont plus rares, affectant environ un sur 1 000 de la population générale.
Signes et symptômes
Physique
Les adénomes hypophysaires sécrétant des hormones provoquent l'une des nombreuses formes d' hyperpituitarisme . Les spécificités dépendent du type d'hormone. Certaines tumeurs sécrètent plus d'une hormone, la combinaison la plus courante étant la GH et la prolactine , qui se traduisent par une croissance osseuse inattendue et une lactation inattendue (chez les hommes et les femmes).
Un patient atteint d'adénome hypophysaire peut présenter des anomalies du champ visuel , classiquement une hémianopsie bitemporale . Elle résulte de la compression du nerf optique par la tumeur. La zone spécifique de la voie visuelle où la compression par ces tumeurs se produit est au niveau du chiasma optique . L'anatomie de cette structure provoque une pression sur elle pour produire un défaut dans le champ visuel temporal des deux côtés, une condition appelée hémianopsie bitemporale . S'il a son origine au- dessus du chiasma optique , le plus souvent dans un craniopharyngiome de la tige pituitaire , le défaut du champ visuel apparaîtra d'abord comme une quadrantanopie inférieure bitemporale , s'il a son origine en dessous du chiasma optique , le défaut du champ visuel apparaîtra d'abord comme un quadrantanopie supérieure bitemporale . L'expansion latérale d'un adénome hypophysaire peut également comprimer le nerf abducens , provoquant une paralysie du droit latéral .
En outre, un adénome hypophysaire peut provoquer des symptômes d' augmentation de la pression intracrânienne . Prolactinomes commencent souvent à donner des symptômes en particulier pendant la grossesse, lorsque l'augmentation de l' hormone niveau oestrogène peut augmenter le taux de croissance de la tumeur.
Divers types de maux de tête sont fréquents chez les patients atteints d'adénome hypophysaire. L'adénome peut être le principal facteur causal du mal de tête ou peut servir à exacerber un mal de tête causé par d'autres facteurs. Parmi les types de maux de tête ressentis figurent à la fois la migraine chronique et épisodique et, plus rarement, divers maux de tête unilatéraux ; Céphalée primaire en coup de couteau, céphalées neuralgiformes unilatérales de courte durée avec injection et larmoiement conjonctivales (SUNCT) - un autre type de céphalée lancinante caractérisée par de courts coups de douleur -, céphalée en grappe et hemicrania continua (HS).
Les symptômes compressifs des adénomes hypophysaires (déficits du champ visuel, diminution de l'acuité visuelle, maux de tête) sont plus fréquemment observés avec les macroadénomes (qui mesurent plus de 10 mm de diamètre) qu'avec les microadénomes (qui mesurent moins de 10 mm de diamètre).
Les adénomes non sécrétants peuvent passer inaperçus pendant une période prolongée car aucune anomalie évidente n'est observée ; la réduction progressive des activités normales due à la diminution de la production d'hormones est plutôt moins évidente. Par exemple, une quantité insuffisante d'hormone adrénocorticotrope signifie que les glandes surrénales ne produisent pas suffisamment de cortisol , ce qui entraîne une récupération lente de la maladie, de l'inflammation et de la fatigue chronique ; l'insuffisance d'hormone de croissance chez l'enfant et l'adolescent entraîne une diminution de la stature mais qui peut avoir bien d'autres explications.
Psychiatrique
Diverses manifestations psychiatriques ont été associées à des troubles hypophysaires, notamment des adénomes hypophysaires. Des symptômes psychiatriques tels que la dépression, l'apathie anxieuse, l'instabilité émotionnelle, l'irritabilité facile et l'hostilité ont été notés.
Complications

- L'acromégalie est un syndrome qui se produit lorsque l' hypophyse antérieure produit un excès d' hormone de croissance (GH). Environ 90 à 95 % des cas d'acromégalie sont causés par un adénome hypophysaire et affectent le plus souvent les adultes d'âge moyen. L'acromégalie peut entraîner une défiguration grave, des complications graves et une mort prématurée si elle n'est pas contrôlée. La maladie, qui est souvent également associée au gigantisme , est difficile à diagnostiquer à un stade précoce et est souvent ignorée pendant de nombreuses années, jusqu'à ce que des modifications des caractéristiques externes, en particulier du visage, deviennent perceptibles avec le temps médian entre le développement des premiers symptômes et le diagnostic étant de douze ans.
- Le syndrome de Cushing est un trouble hormonal qui provoque l'hypercortisolisme, c'est-à-dire des niveaux élevés de cortisol dans le sang. La maladie de Cushing (MC) est la cause la plus fréquente du syndrome de Cushing, responsable d'environ 70 % des cas. La MC survient lorsqu'un adénome hypophysaire provoque une sécrétion excessive d' hormone adrénocorticotrope (ACTH) qui stimule les glandes surrénales à produire des quantités excessives de cortisol .
- La maladie de Cushing peut provoquer de la fatigue, une prise de poids, des amas graisseux autour de l'abdomen et du bas du dos (obésité du tronc) et du visage ("visage lunaire"), des vergetures ( striae ) sur la peau de l'abdomen, des cuisses, des seins et des bras, de l' hypertension , l'intolérance au glucose et diverses infections. Chez la femme, il peut provoquer une croissance excessive de la pilosité faciale ( hirsutisme ) et chez l'homme une dysfonction érectile . Les manifestations psychiatriques peuvent inclure la dépression, l' anxiété , l'irritabilité facile et l'instabilité émotionnelle. Elle peut également entraîner diverses difficultés cognitives .
- L'hyperpituitarisme est une maladie du lobe antérieur de l'hypophyse qui est généralement causée par un adénome hypophysaire fonctionnel et entraîne une hypersécrétion d'hormones adénohypophysaires telles que l'hormone de croissance; prolactine; thyrotropine; hormone lutéinisante; hormone de stimulation de follicule; et l'hormone adrénocorticotrope.
- L'apoplexie hypophysaire est une affection qui survient lorsque les adénomes hypophysaires présentent soudainement une hémorragie interne, provoquant une augmentation rapide de la taille ou lorsque la tumeur dépasse son apport sanguin, ce qui provoque une nécrose des tissus et un gonflement ultérieur des tissus morts. L'apoplexie hypophysaire se présente souvent avec une perte de vision et des maux de tête d'apparition soudaine et nécessite un traitement rapide souvent avec des corticostéroïdes et si nécessaire une intervention chirurgicale.
- Le diabète insipide central est causé par une diminution de la production de l' hormone antidiurétique vasopressine qui provoque une soif intense et une production excessive d'urines très diluées ( polyurie ) pouvant entraîner une déshydratation . La vasopressine est produite dans l' hypothalamus et est ensuite transportée le long de la tige pituitaire et stockée dans le lobe postérieur de la glande pituitaire qui la sécrète ensuite dans la circulation sanguine.
Comme l'hypophyse est à proximité immédiate du cerveau, les adénomes invasifs peuvent envahir la dure-mère , l' os crânien ou l'os sphénoïde .
Facteurs de risque
Néoplasie endocrinienne multiple
Les adénomes de l'hypophyse antérieure sont une caractéristique clinique majeure de la néoplasie endocrinienne multiple de type 1 (NEM1), un syndrome endocrinien héréditaire rare qui affecte 1 personne sur 30 000. La NEM provoque diverses combinaisons de tumeurs bénignes ou malignes dans diverses glandes du système endocrinien ou peut entraîner une hypertrophie des glandes sans formation de tumeurs. Elle affecte souvent les glandes parathyroïdes , les cellules des îlots pancréatiques et le lobe antérieur de l'hypophyse. NEM1 peut également provoquer des tumeurs non endocriniennes telles que des angiofibromes faciaux , des collagénomes , des lipomes , des méningiomes , des épendymomes et des léiomyomes . Environ 25 % des patients atteints de NEM1 développent des adénomes hypophysaires.
Complexe de Carney
Le complexe de Carney (CNC), également connu sous le nom de syndrome de LAMB et de syndrome de NAME, est une maladie autosomique dominante comprenant des myxomes du cœur et de la peau, une hyperpigmentation de la peau ( lentiginose ) et une hyperactivité endocrinienne et est distincte de la triade de Carney . Environ 7 % de tous les myxomes cardiaques sont associés au complexe de Carney. Les patients atteints de CNC développent des tumeurs hypophysaires produisant de l' hormone de croissance (GH) et, dans certains cas, ces mêmes tumeurs sécrètent également de la prolactine . Il n'existe cependant pas de prolactinomes isolés ou de tout autre type de tumeur hypophysaire. Chez certains patients atteints de CNC, l'hypophyse est caractérisée par des zones hyperplasiques avec l'hyperplasie précédant très probablement la formation d'adénomes producteurs de GH.
Adénome hypophysaire familial isolé
L'adénome hypophysaire isolé familial (APIF) est un terme utilisé pour identifier une affection qui présente une transmission autosomique dominante et qui se caractérise par la présence de deux patients apparentés ou plus affectés par des adénomes de l'hypophyse uniquement, sans aucun autre symptôme associé qui surviennent dans les néoplasies endocriniennes multiples de type 1 (NEM-1) ou le complexe de Carney . Le FIPA a été décrit pour la première fois dans une cohorte limitée de familles par le groupe Albert Beckers à Liège, en Belgique ; plus tard FIPA a été entièrement caractérisé dans une étude internationale multicentrique de 64 familles. Les familles FIPA sont divisées en familles homogènes et présentent le même type d'adénome hypophysaire chez tous les membres de la famille touchés (par exemple, uniquement l' acromégalie , uniquement le prolactinome , etc.), tandis que les familles FIPA hétérogènes peuvent avoir différents adénomes hypophysaires chez les membres de la famille touchés.
Génétique du FIPA
FIPA a deux causes génétiques connues, des mutations dans le gène de la protéine interagissant avec le récepteur AH (AIP) et des duplications dans le chromosome Xq26.3 qui incluent le gène GPR101 qui provoque également le syndrome d'acrogigantisme lié à l'X (X-LAG). Environ 15 à 20 % des familles FIPA sont porteuses d'une mutation ou délétion du gène AIP de la lignée germinale , et la maladie survient sur un mode autosomique dominant avec une pénétrance incomplète, ce qui signifie qu'environ 20 % des porteurs de la mutation AIP développeront un adénome hypophysaire. Les adénomes hypophysaires associés à une mutation AIP (présentant soit comme FIPA, soit comme des cas individuels non familiaux) sont généralement des adénomes sécrétant de l'hormone de croissance ( acromégalie ) ou de la prolactine ( prolactinome ) qui sont de grande taille (macroadénomes) et surviennent souvent chez les enfants, les adolescents et les jeunes adultes. Daly et ses collègues ont montré que les cas d'acromégalie avec mutations AIP se sont produits environ 20 ans avant les cas d'acromégalie sans mutations AIP et que ces tumeurs sont volumineuses et relativement résistantes au traitement. En raison de leur jeune âge d'apparition, les mutations AIP sont la cause génétique la plus fréquente du gigantisme hypophysaire (29 % des cas).
X-LAG est un syndrome rare de tumeurs/hyperplasie hypophysaires d'apparition très précoce qui entraîne un excès d'hormone de croissance et une prolifération sévère et un gigantisme hypophysaire. Trois familles FIPA avec X-LAG ont été signalées à ce jour, toutes ayant une transmission d'une duplication du chromosome Xq26.3 de la mère affectée au fils affecté. Les caractéristiques de la maladie d'un gigantisme hypophysaire d' apparition très jeune entraînent une prolifération sévère si elle n'est pas traitée de manière adéquate ; bon nombre des humains les plus grands de l'histoire (par exemple, Robert Pershing Wadlow ; Sandy Allen , André Rousimoff (André le Géant), Zeng Jinlian ) avaient une histoire clinique similaire à celle des patients atteints du syndrome X-LAG. L'individu historique le plus grand avec une cause génétique connue était Julius Koch (Geant Constantin) qui s'est avéré avoir X-LAG sur l'étude génétique de son squelette. X-LAG a une pénétrance de 100 % jusqu'à présent (tous atteints de la duplication Xq26.3 ont la maladie et cela affecte principalement les femmes. Les cas isolés non familiaux de X-LAG peuvent avoir une duplication constitutionnelle d'un chromosome Xq26.3 comprenant GPR101 , ou mosaïcisme pour la duplication (présent dans une minorité de cellules) dans le cas de patients masculins isolés.X-LAG provoque environ 10% des cas de gigantisme hypophysaire .
Mécanisme

L'hypophyse ou l'hypophyse est souvent appelée la "glande maîtresse" du corps humain. Faisant partie de l' axe hypothalamo-hypophysaire , il contrôle la plupart des fonctions endocriniennes du corps via la sécrétion de diverses hormones dans le système circulatoire . L'hypophyse est située sous le cerveau dans une dépression ( fosse ) de l' os sphénoïde connue sous le nom de selle turcique . Bien que anatomiquement et fonctionnellement connectée au cerveau, l'hypophyse se trouve à l'extérieur de la barrière hémato-encéphalique . Il est séparé de l' espace sous - arachnoïdien par le diaphragme de la selle , donc l' arachnoïde-mère et donc le liquide céphalo-rachidien ne peuvent pas pénétrer dans la selle turcique.
L'hypophyse est divisée en deux lobes, le lobe antérieur (qui représente les deux tiers du volume de la glande), et le lobe postérieur (un tiers du volume) séparés par la pars intermedia .
Le lobe postérieur (le lobe neural ou neurohypophyse) de l'hypophyse n'est pas, malgré son nom, une véritable glande . Le lobe postérieur contient des axones de neurones qui s'étendent de l' hypothalamus auquel il est connecté via la tige pituitaire. Les hormones vasopressine et ocytocine , produites par les neurones des noyaux supraoptique et paraventriculaire de l'hypothalamus, sont stockées dans le lobe postérieur et libérées par les terminaisons axonales ( dendrites ) à l'intérieur du lobe.
Le lobe antérieur de l' hypophyse (adénohypophyse) est une véritable glande qui produit et sécrète six hormones différentes : hormone thyréostimulante (TSH), hormone adrénocorticotrope (ACTH), hormone folliculo-stimulante (FSH), hormone lutéinisante (LH), hormone de croissance (GH) et la prolactine (PRL).
Diagnostic
Le diagnostic d'adénome hypophysaire peut être posé, ou du moins suspecté, par une constellation de symptômes apparentés présentés ci-dessus.
Le diagnostic différentiel inclut le tuberculome hypophysaire, en particulier dans les pays en développement et chez les patients immunodéprimés. Le diagnostic est confirmé par le test des taux d'hormones et par l'imagerie radiographique de l'hypophyse (par exemple, par tomodensitométrie ou IRM ).
Classification
Contrairement aux tumeurs de l'hypophyse postérieure, les adénomes hypophysaires sont classés comme des tumeurs endocrines (pas des tumeurs cérébrales). Les adénomes hypophysaires sont classés en fonction de critères anatomiques , histologiques et fonctionnels.
- Les tumeurs anatomiquement hypophysaires sont classées selon leur taille en fonction des résultats radiologiques ; soit des microadénomes (inférieurs à < 10 mm) soit des macroadénomes (égales ou supérieures à ≥ 10 mm).
- La classification basée sur les résultats radio-anatomiques place les adénomes en 1 des 4 grades (I-IV) :
- Stade I : microadénomes (<1 cm) sans expansion de la selle.
- Stade II : macroadénomes (≥1 cm) et pouvant s'étendre au-dessus de la selle.
- Stade III : macroadénomes avec élargissement et envahissement du plancher ou extension suprasellaire.
- Le stade IV est la destruction de la selle.
- La classification histologique utilise une caractérisation immunohistologique des tumeurs en fonction de leur production hormonale. Historiquement , ils ont été classés comme étant soit basophiles , acidophiles ou chromophobes sur la base de si oui ou non ils ont pris la tinctoriale taches hématoxyline et éosine . Cette classification est tombée en désuétude, au profit d'une classification basée sur le type d' hormone sécrétée par la tumeur. Environ 20 à 25 % des adénomes ne sécrètent aucune hormone active facilement identifiable (« tumeurs non fonctionnelles »), mais ils sont encore parfois appelés « chromophobes ».
- La classification fonctionnelle est basée sur l'activité endocrinienne des tumeurs telle que déterminée par les taux d' hormones sériques et la sécrétion d'hormones cellulaires dans le tissu hypophysaire détectées par coloration immunohistochimique . Les valeurs du « pourcentage de cas de production d'hormones » sont les fractions d'adénomes produisant chaque hormone apparentée de chaque type de tumeur par rapport à tous les cas de tumeurs hypophysaires, et ne sont pas directement corrélées aux pourcentages de chaque type de tumeur en raison d'incidences plus ou moins importantes de absence de sécrétion de l'hormone attendue. Ainsi, les adénomes non sécrétoires peuvent être soit des adénomes à cellules nulles, soit un adénome plus spécifique qui reste cependant non sécrétif.
- Tout type d'adénocarcinome hypophysaire répertorié dans le tableau ci-dessous peut provoquer des symptômes compressifs dus à une expansion locale en plus des effets systémiques des hormones sécrétées répertoriées dans la colonne pathologie.
- Les adénomes à cellules nulles, par définition, ne sécrètent pas d'hormones, mais ils provoquent généralement des effets compressifs sur la tige pituitaire (effet de tige). Cela conduit à une diminution des niveaux de dopamine de l'hypothalamus atteignant l'hypophyse antérieure. La dopamine exerce un effet inhibiteur sur la sécrétion de prolactine. En l'absence de cet effet inhibiteur, les taux de prolactine augmentent et sont souvent augmentés dans les adénomes à cellules nulles. Cela conduit à des symptômes d'hypogonadisme.
| Type d'adénome | Sécrétion | coloration | Pathologie | Pourcentage de cas de production d'hormones | Pourcentage de cas silencieux |
|---|---|---|---|---|---|
| adénomes lactotrophes ( prolactinomes ) | sécréter de la prolactine | acidophile | galactorrhée , hypogonadisme , aménorrhée , infertilité et impuissance | 30% | <9% |
| adénomes somatotrophes | sécréter l'hormone de croissance (GH) | acidophile | acromégalie chez l'adulte; gigantisme chez les enfants | 15% | <9% |
| adénomes corticotrophes | sécrètent l' hormone adénocorticotrope (ACTH) | basophile | La maladie de Cushing | 2-6% | dix% |
| adénomes gonadotrophiques | sécrètent l'hormone lutéinisante (LH), l'hormone folliculo-stimulante (FSH) et leurs sous-unités | basophile | ne provoquent généralement pas de symptômes, parfois hypergonadisme | dix% | 73% |
| adénomes thyrotrophes (rares) | sécréter la thyréostimuline (TSH) | basophile à chromophobe | parfois hyperthyroïdie , ne provoque généralement pas de symptômes | Moins que 1% | <9% |
| adénomes à cellules nulles | ne sécrètent pas d'hormones | peut se colorer positivement pour la synaptophysine | Asymptomatique ou hypogonadisme | 25 % des adénomes hypophysaires sont non sécrétants | 1% |
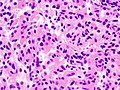
Adénome hypophysaire somatotrophe, montrant un cytoplasme acidophile
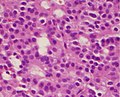
Un adénome hypophysaire gonadotrophe silencieux qui est, dans ce cas, éosinophile (contrairement aux cellules normales, basophiles, gonadotrophes)
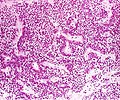
Les vrais adénomes à cellules nulles sont généralement composés de cellules uniformes légèrement atypiques avec un cytoplasme chromophobe. Ce cas a une architecture papillaire similaire aux adénomes gonadotrophiques.
_GH_production.jpg)


Incidentalomes hypophysaires
Les incidentalomes hypophysaires sont des tumeurs hypophysaires caractérisées comme une découverte fortuite . Ils sont souvent découverts par tomodensitométrie (TDM) ou imagerie par résonance magnétique (IRM), réalisée dans l'évaluation de conditions médicales non liées telles qu'un traumatisme crânien suspecté , dans la stadification du cancer ou dans l'évaluation de symptômes non spécifiques tels que les vertiges et les maux de tête . Il n'est pas rare qu'elles soient découvertes à l' autopsie . Dans une méta-analyse , des adénomes ont été trouvés dans une moyenne de 16,7 % dans les études post mortem, la plupart étant des microadénomes (<10 mm) ; les macrodénomes ne représentaient que 0,16% à 0,2% des personnes décédées. Bien que les microadénomes hypophysaires non sécrétants et non invasifs soient généralement considérés comme étant littéralement et cliniquement bénins , il existe à ce jour peu d'études de faible qualité pour étayer cette affirmation.
Il a été recommandé dans les directives de pratique clinique actuelles (2011) de l' Endocrine Society - une organisation médicale internationale professionnelle dans le domaine de l'endocrinologie et du métabolisme - que tous les patients atteints d'incidentalomes hypophysaires subissent une anamnèse complète et un examen physique , des évaluations de laboratoire pour dépistage de l'hypersécrétion hormonale et de l' hypopituitarisme . Si la lésion est à proximité immédiate des nerfs optiques ou du chiasma optique , un examen du champ visuel doit être effectué. Pour ceux qui ont des incidentalomes qui ne nécessitent pas d'ablation chirurgicale, des évaluations cliniques de suivi et une neuroimagerie doivent être effectuées ainsi que des examens du champ visuel de suivi pour les incidentalomes qui jouxtent ou compriment le nerf optique et le chiasma et des tests endocriniens de suivi pour les macroincidentalomes.
Adénome hypophysaire ectopique
Un adénome hypophysaire ectopique (survenant à un endroit anormal) est un type rare de tumeur qui survient en dehors de la selle turcique , le plus souvent dans le sinus sphénoïde , la région suprasellaire, le nasopharynx et les sinus caverneux .
Métastases à l'hypophyse
Les carcinomes qui métastasent dans l'hypophyse sont rares et généralement observés chez les personnes âgées, les cancers du poumon et du sein étant les plus fréquents. Chez les patientes atteintes d'un cancer du sein, des métastases de l'hypophyse surviennent dans environ 6 à 8 % des cas.
Les métastases hypophysaires symptomatiques ne représentent que 7 % des cas rapportés. Chez les personnes symptomatiques, le diabète insipide survient souvent avec des taux d'environ 29 à 71 %. D'autres symptômes fréquemment rapportés incluent un dysfonctionnement de l'hypophyse antérieure, des anomalies du champ visuel, des maux de tête/douleur et une ophtalmoplégie .
Traitement
Les options de traitement dépendent du type de tumeur et de sa taille :
- Les prolactinomes sont le plus souvent traités avec de la cabergoline ou du quinagolide (tous deux des agonistes de la dopamine ), qui diminuent la taille de la tumeuret atténuent lessymptômes, suivis d'une imagerie en série pour détecter toute augmentation de taille. Le traitement, lorsque la tumeur est volumineuse, peut se faire par radiothérapie, protonthérapie ou chirurgie, et les patients répondent généralement bien. Contrairement aux prolactinomes, les adénomes thyrotrophiques répondent généralement mal au traitement par agoniste dopaminergique.
- Les adénomes somatotrophes répondent à l' octréotide ou au lanréotide , qui sont des analogues de la somatostatine à action prolongée . Ces analogues du récepteur de la somatostatine inhibent la sécrétion de l'hormone de croissance. Ils se sont avérés efficaces d'environ 50 à 55 % pour réduire la masse tumorale et réduire les niveaux d' hormone de croissance et de facteur de croissance 1 ( IGF-1 ) semblable à l'insuline dans les études. L'antagoniste des récepteurs de l'hormone de croissance pegvisomant est également utilisé dans le traitement des adénomes somatotrophes. Le pegvisomant bloque l'action de l'hormone de croissance. Il peut être utilisé en monothérapie ou associé à un analogue de la somatostatine.
- La chirurgie est un traitement courant pour les tumeurs hypophysaires. L'approche normale est l'adénectomie trans-sphénoïdale , qui permet généralement d'enlever la tumeur sans affecter le cerveau ou les nerfs optiques.
- La radiothérapie est également utilisée pour traiter les adénomes hypophysaires. Les exemples incluent la radiothérapie par faisceau externe ou par faisceau de protons ou la radiochirurgie stéréotaxique. L'irradiation externe des adénomes hypophysaires peut arrêter la croissance tumorale pendant plusieurs années, mais l'insuffisance hypophysaire se développe dans les 10 ans chez la plupart des patients nécessitant un remplacement hormonal à vie. La radiothérapie pour les adénomes hypophysaires est associée à une multiplication par 4 de la mortalité due aux maladies cérébrovasculaires.
Voir également
Les références
Liens externes
| Classification | |
|---|---|
| Ressources externes |