
Migalastat - Migalastat
 | |
| Donnée clinique | |
|---|---|
| Prononciation | mi GAL une stat |
| Appellations commerciales | Galafold |
| Autres noms | DDIG, AT1001, 1-désoxygalactonojirimycine |
| AHFS / Drugs.com | Monographie |
| Données de licence | |
Catégorie grossesse |
|
| Voies administratives |
Par voie orale ( gélules ) |
| code ATC | |
| Statut légal | |
| Statut légal | |
| Données pharmacocinétiques | |
| Biodisponibilité | 75% |
| Liaison protéique | Rien |
| Métabolites | O - glucuronides (<15%) |
| Demi-vie d' élimination | 3 à 5 heures (dose unique) |
| Excrétion | Urine (77 %), fèces (20 %) |
| Identifiants | |
| |
| Numero CAS | |
| CID PubChem | |
| Banque de médicaments | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Données chimiques et physiques | |
| Formule | C 6 H 13 N O 4 |
| Masse molaire | 163,173 g·mol -1 |
| Modèle 3D ( JSmol ) | |
| |
| |
Le migalastat , vendu sous le nom de marque Galafold , est un médicament destiné au traitement de la maladie de Fabry , une maladie génétique rare. Il a été développé par Amicus Therapeutics . La Food and Drug Administration (FDA) des États-Unis lui a accordé le statut de médicament orphelin en 2004, et la Commission européenne a suivi en 2006. Le Comité des médicaments à usage humain (CHMP) de l' Agence européenne des médicaments a accordé au médicament une autorisation de mise sur le marché sous le nom de Galafold en Mai 2016.
La Food and Drug Administration (FDA) des États-Unis considère qu'il s'agit d'un médicament de premier ordre .
Utilisations médicales
Le migalastat est utilisé pour le traitement à long terme de la maladie de Fabry chez les adultes et les adolescents âgés de 16 ans ou plus présentant une mutation sensible de l'enzyme alpha-galactosidase A (α-GalA). Une mutation « acceptable » est une mutation qui conduit à un mauvais repliement de l'enzyme, mais n'altèrerait pas autrement de manière significative sa fonction.
Sur la base d'un test in vitro , Amicus Therapeutics a publié une liste de 269 mutations amenables et près de 600 mutations non amenables. Environ 35 à 50 % des personnes atteintes de Fabry ont une mutation favorable.
Effets indésirables
L'effet secondaire le plus courant dans les essais cliniques était le mal de tête (chez environ 10 % des personnes qui en prenaient). Les effets secondaires moins fréquents (entre 1 et 10 % des personnes) comprenaient des symptômes non spécifiques tels que des étourdissements, de la fatigue et des nausées , mais aussi une dépression. Les effets secondaires rares possibles n'ont pas pu être évalués en raison du faible nombre de sujets dans les essais cliniques dans lesquels les effets indésirables ont été mesurés.
Interactions
Lorsqu'il est associé à l' agalsidase alpha ou bêta intraveineuse , qui sont des versions recombinantes de l'enzyme -GalA, le migalastat augmente les concentrations tissulaires de α-GalA fonctionnelle par rapport à l'agalsidase administrée seule. C'est un effet attendu et souhaité.
Le migalastat n'inhibe ni n'induit les enzymes hépatiques du cytochrome P450 ni les protéines de transport et devrait donc présenter un faible potentiel d'interactions avec d'autres médicaments.
Pharmacologie
Mécanisme d'action
La maladie de Fabry est une maladie génétique causée par diverses mutations de l'enzyme α-GalA, responsable de la dégradation du sphingolipide globotriaosylcéramide (Gb3), parmi d'autres glycolipides et glycoprotéines . Certaines de ces mutations entraînent un mauvais repliement de la -GalA, qui échoue par la suite au contrôle de la qualité des protéines dans le réticulum endoplasmique et se décompose. Le manque de α-GalA fonctionnelle entraîne une accumulation de Gb3 dans les vaisseaux sanguins et d'autres tissus, avec un large éventail de symptômes, notamment des problèmes rénaux, cardiaques et cutanés.
Le migalastat est un puissant inhibiteur de la -GalA disponible par voie orale ( IC 50 : 4 M ). Lorsqu'il se lie à une α-GalA défectueuse, il déplace le comportement de repliement vers la conformation appropriée, résultant en une enzyme fonctionnelle à condition que la mutation soit propice. Les molécules dotées de ce type de mécanisme sont appelées chaperons pharmacologiques .
Lorsque l'enzyme atteint sa destination, le lysosome , le migalastat se dissocie en raison du faible pH et de l'abondance relative de Gb3 et d'autres substrats , laissant -GalA libre de remplir sa fonction. Selon la mutation, l' EC 50 est comprise entre 0,8 µM et plus de 1 mM dans les modèles cellulaires.

L'enzyme alpha-galactosidase A (α-GalA)

Le globotriaosylcéramide (Gb3), un substrat de l'α-GalA, a un D - galactose terminal structurellement similaire au migalastat.
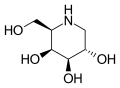
Migalastat (vue "de haut")



Pharmacocinétique
Le migalastat est presque complètement absorbé par l'intestin; la prise du médicament avec de la nourriture diminue son absorption d'environ 40 %. La biodisponibilité totale est d'environ 75 % lorsqu'elle est prise sans nourriture. La substance n'est pas liée aux protéines du plasma sanguin .
Seule une petite fraction d'une dose de migalastat est métabolisée, principalement en trois O - glucuronides déshydrogénés (4 % de la dose) et un certain nombre de métabolites non spécifiés (10 %). Le médicament est principalement éliminé par les urines (77 %) et dans une moindre mesure par les selles (20 %). Pratiquement tous les métabolites sont excrétés dans l'urine. La demi-vie d'élimination est de trois à cinq heures après une dose unique.
Chimie

Le migalastat est utilisé sous forme de chlorhydrate , qui est un solide cristallin blanc et soluble dans l'eau. La molécule a quatre atomes de carbone asymétriques avec la même stéréochimie que le sucre D - galactose , mais il manque le premier groupe hydroxyle . Il a un atome d'azote dans le cycle au lieu d'un oxygène, ce qui en fait un iminosucre .
La structure est formellement dérivée de la nojirimycine .
Histoire
Le migalastat a été isolé en tant que produit de fermentation de la bactérie Streptomyces lydicus ( souche PA-5726) en 1988 et appelé 1-désoxygalactonojirimycine . En 2004, il a été désigné médicament orphelin par la FDA américaine pour le traitement de la maladie de Fabry, et en 2006, le CHMP européen a fait de même. Le parrainage du médicament a été transféré à plusieurs reprises au cours des années suivantes : d'Amicus Therapeutics à Shire Pharmaceuticals en 2008, de nouveau à Amicus en 2010, à Glaxo en 2011, et à nouveau à Amicus en 2014.
Deux essais cliniques de phase III avec un total d'environ 110 sujets ont été menés entre 2009 et 2015, un en double aveugle comparant le médicament à un placebo , et un le comparant à la α-GalA recombinante sans aveugle. Le migalastat a stabilisé la fonction cardiaque et rénale au cours de la période de 30 mois de ces essais.
En septembre 2015, Amicus a annoncé qu'elle soumettrait une demande de nouveau médicament (NDA) pour l' approbation accélérée du migalastat à la FDA d'ici la fin 2015. Le CHMP a recommandé l'approbation en avril 2016, mais la FDA a rejeté la demande en novembre pour avoir données en novembre 2016. Le médicament a été approuvé dans l'Union européenne en mai 2016. L'Allemagne a été le premier pays où le migalastat a été lancé. Après que Scott Gottlieb soit devenu commissaire de la FDA en 2017, le PDG d'Amicus a commencé à le faire pression directement pour que la FDA accepte la NDA et en février 2018, la FDA l'a acceptée et a promis une réponse d'ici août 2018.
Voir également
- Miglustat , un médicament pour le traitement de la maladie de Gaucher, avec une structure similaire
- 1-Déoxynojirimycine , un stéréoisomère du migalastat
Les références
Liens externes
- "Migalastat" . Portail d'information sur les médicaments . Bibliothèque nationale de médecine des États-Unis.